
1 Form Proposal - Invoice 1.4 serial key or number

1 Form Proposal - Invoice 1.4 serial key or number
Guidance Document For Clinical Trial Sponsors: Clinical Trial Applications
(PDF Version - 188 K)
Our file number: 13-108409-403
Guidance Document for Clinical Trial Sponsors: Clinical Trial Applications
Health Canada is pleased to announce the release of the finalizedGuidance Document for Clinical Trial Sponsors: Clinical Trial Applications which provides guidance to all sponsors [for example (e.g.) industry, academic, contract research organization] seeking authorization to sell or import a drug for the purpose of a clinical trial in Canada. The Guidance was revised based on stakeholder consultation processes and as part of Health Canada's statutory review of Part C, Division 5 of the Food and Drug Regulations (Drugs for Clinical Trials Involving Human Subjects).
The Guidance for Clinical Trial Applications (CTAs) is consistent with the new Common Technical Document (CTD) format and is clear on application requirements. This Guidance document supersedes the previous Guidance for Clinical Trial Sponsors: Clinical Trial Applications (June 25, 2003). The revised guidance includes application requirements for comparative bioavailability trials and filing requirements for the importation of clinical trial supplies. It includes clarifications to amendment and notification requirements, study termination and closure criteria, application and review processes, and adverse drug reaction reporting criteria as well as format requirements. All stakeholder comments were considered in the finalization of this guidance document. A table of comments from industry stakeholders in response to the draft CTA Guidance is available upon request.
As a reminder, Health Canada is advising sponsors of the new electronic adverse drug reactions (ADR) reporting that is currently in pilot with some sponsors. Those sponsors who have an established connection with the Canada Vigilance Production stream should submit their reports only to the appropriate Directorates: Therapeutic Products Directorate (TPD), Biologics and Genetic Therapies Directorate (BGTD) or Marketed Health Products Directorate (MHPD) [that is (i.e.) a report no longer needs to be sent in duplicate to multiple directorates]. For the sponsors who have not yet established this connection, they should continue submitting their reports by fax or by courier. The Health Canada website will provide further clarification on Health Canada's ADR reporting requirements.
Improving the quality of CTAs that are submitted by sponsors and gaining efficiencies with CTA screening, regulatory review and safety reporting, will provide benefits to those involved in the conduct of clinical trials, and most of all to Canadians.
Questions or comments regarding this initiative should be directed to:
Office of Clinical Trials
Therapeutic Products Directorate
Health Canada
1600 Scott Street
Holland Cross, Tower B
5th Floor, Address Locator 3105A
Ottawa, Ontario
K1A 0K9
Facsimile: 613-954-4474
E-mail: OCT_BEC_Enquiries@hc-sc.gc.ca
Clinical Trial Applications
Published by authority of the Minister of Health
Date Adopted: 2003/06/25
Revised Date: 2011/11/07
Effective Date: 2013/05/29
Revised Date: 2016/03/17
Guidance documents are meant to provide assistance to industry and health care professionals on how to comply with governing statutes and regulations. Guidance documents also provide assistance to staff on how Health Canada mandates and objectives should be implemented in a manner that is fair, consistent and effective.
Guidance documents are administrative instruments not having force of law and, as such, allow for flexibility in approach. Alternate approaches to the principles and practices described in this document may be acceptable provided they are supported by adequate justification. Alternate approaches should be discussed in advance with the relevant program area to avoid the possible finding that applicable statutory or regulatory requirements have not been met.
As a corollary to the above, it is equally important to note that Health Canada reserves the right to request information or material, or define conditions not specifically described in this document, in order to allow the Department to adequately assess the safety, efficacy or quality of a therapeutic product. Health Canada is committed to ensuring that such requests are justifiable and that decisions are clearly documented.
This document should be read in conjunction with the accompanying notice and the relevant sections of other applicable Guidance documents.
| Change Date | Location (section, paragraph) | Nature of and/or Reason for Change |
|---|---|---|
| 2003/06/11 | Date of Original Adoption | |
| 2008/02/27 | Appendix 4 and throughout document | Reflecting necessary changes from finalization of the protocol safety and efficacy assessment template - clinical trial application [Protocol Safety and Efficacy Assessment Template - Clinical Trial Application (PSEAT-CTA)]. |
| 2009/03/12 | Section 2.3 | Administrative changes. |
| 2011/11/07 | Section 2.7.5, Appendix 4 and throughout document to address stakeholder feedback | New requirements on the Importation of Clinical Trial Drugs and accompanying form. |
| 2016/03/17 | Throughout document | Change in requirements for electronic specifications |
The Food and Drugs Act and the Food and Drug Regulations (herein referred to as the Regulations) govern the sale and importation of drugs for use in human clinical trials in Canada. This document provides guidance on the regulatory obligations pursuant to Part C, Division 5 of the Regulations, Drugs for Clinical Trials Involving Human Subjects.
To provide sponsors seeking authorization to conduct a clinical trial in Canada with guidance to support the protection of clinical trial subjects and contributes to the high standards of excellence in research and development in Canada. This document clarifies application and post-authorization requirements and outlines procedures for obtaining authorization.
With the exception of Phase IV studies, clinical trial sponsors must submit a clinical trial application (CTA) to Health Canada for authorization to sell or import a drug for the purpose of a clinical trial.
Clinical trial sponsors must conduct clinical trials according to generally accepted principles of good clinical practice that ensure the protection of the rights, safety and well-being of clinical trial subjects and other persons.
Research Ethics Boards (REBs) have an important role in the oversight of the conduct of clinical trials. Sponsors are required by the Regulations to obtain REB approval for each clinical trial site prior to commencing the trial at that site [C.05.006(1)(c)].
The Regulations are generally consistent with the principles, definitions and standards found in the International Conference on Harmonisation (ICH) Guidance documents on clinical trials. Where inconsistencies exist, the Regulations take precedence.
The format for CTAs as outlined in this Guidance document is consistent with that used for other types of drug submissions filed to Health Canada, based on the format of the ICH Common Technical Document (CTD). Although the scope of the ICH CTD does not include applications at the clinical research stage of development, the modular format of the CTD is being extended to CTAs to facilitate the preparation of drug submission information throughout the lifecycle of a drug.
1.3 Scope and Application
The information provided in this Guidance document is for clinical trials involving drugs (pharmaceuticals and/or biologics and radiopharmaceuticals) in human subjects. In the Regulations a clinical trial is an investigation in respect of a drug that is intended to discover or verify the clinical, pharmacological or pharmacodynamic effects of the drug, identify any adverse events in respect of the drug, study the absorption, distribution, metabolism and excretion of the drug, or ascertain the safety or efficacy of the drug.
This Guidance document applies to all sponsors (e.g., industry, academic, contract research organization, etc.) conducting clinical trials, as specified below:
- Clinical trials of a product that is not authorized for sale in Canada including clinical trials for Phases I through III of drug development and comparative bioavailability studies.
- Clinical trials for marketed drugs where the proposed use of the drug is outside the parameters of the Notice of Compliance (NOC) or Drug Identification Number (DIN) application.
- Clinical Trial Application Amendments (CTA-A) [C.05.008] and Notifications (CTA-N) [C.05.007].
This Guidance document does not apply to Phase IV clinical trials or clinical trials involving medical devices or natural health products except where indicated.
This Guidance document supersedes the previous Guidance for Clinical Trial Sponsors: Clinical Trial Applications (June 25, 2003).
The regulatory requirements respecting drugs to be used for the purposes of clinical trials were originally developed in the early 1960s. On September 1, 2001, the regulatory amendments to Part C, Division 5 of the Food and Drug Regulations (Drugs for Clinical Trials Involving Human Subjects) came into force to strengthen protections for clinical trial subjects in Canada.
Health Canada completed a review of the clinical trials regulatory framework through 2006-2008. This review included input received from stakeholders, and consideration of best practices in other countries, as well as Health Canada's experience with the existing regulatory framework (Clinical Trials Regulatory Review: Targeted Measures for a Strengthened Framework). Subsequently, Health Canada proceeded with a series of initiatives, one of which included consultations on the type of guidance required by industry to assist in meeting regulatory obligations (Spring/Summer 2008). During the consultations, stakeholders noted the need for better guidance on processes, requirements, and roles and responsibilities in clinical trial activities.
In response to these consultations, the Guidance for Clinical Trial Sponsors - Clinical Trial Applications has been updated to reflect stakeholder concerns and improve clarity and communication. The guidance includes new information for sponsors regarding information needs and processes related to CTAs, CTA-As and CTA-Ns.
2. Guidance for Implementation
2.1 Abbreviations/Definitions
- ADR
- Adverse Drug Reaction
- BGTD
- Biologics and Genetic Therapies Directorate
- CIOMS
- Council for International Organizations of Medical Sciences
- CR
- Central Registry
- CTA
- Clinical Trial Application
- CTA-A
- Clinical Trial Application-Amendment
- CTA-N
- Clinical Trial Application-Notification
- CTSI
- Clinical Trial Site Information
- CTD
- Common Technical Document
- DIN
- Drug Identification Number
- DMF
- Drug Master File
- GCP
- Good Clinical Practice
- HPFBI
- Health Products and Food Branch Inspectorate
- IB
- Investigator's Brochure
- ICF
- Informed Consent Forms
- ICH
- International Conference on Harmonisation
- ITA
- Investigational Testing Application
- NOC
- Notice of Compliance
- NOL
- No Objection Letter
- NSN
- Not Satisfactory Notice
- PSEAT-CTA
- Protocol Safety and Efficacy Assessment Template - Clinical Trial Application
- QIS
- Quality Information Summary
- QIS-PER
- Quality Information Summary - Positron-Emitting Radiopharmaceuticals
- QIS-R
- Quality Information Summary - Radiopharmaceuticals
- QIU
- Qualified Investigator Undertaking
- QOS
- Quality Overall Summary
- QOS-CE
- Quality Overall Summary - Chemical Entities (Clinical Trial Applications)
- REB
- Research Ethics Board
- TPD
- Therapeutic Products Directorate
Most of the definitions listed below were taken from the Regulations, and the Health Canada / ICH Guidance Documents E6: Guideline for Good Clinical Practice: Consolidated Guideline (ICH E6)and E8: General Considerations for Clinical Trials.
- Adverse Drug Reaction
- Any noxious and unintended response to a drug that is caused by the administration of any dose of the drug.
- Adverse Event
- Any adverse occurrence in the health of a clinical trial subject who is administered a drug, that may or may not be caused by the administration of the drug, and includes an adverse drug reaction.
- Authorized Clinical Trial
- For the purposes of this document, an authorized clinical trial is one that has been filed with Health Canada and has not received an objection from Health Canada within 30 days. Health Canada typically issues a No Objection Letter in instances where no objection has been raised.
- Clinical Trial
- An investigation in respect of a drug for use in humans that involves human subjects and that is intended to discover or verify the clinical, pharmacological or pharmacodynamic effects of the drug, identify any adverse events in respect of the drug, study the absorption, distribution, metabolism and excretion of the drug, or ascertain the safety or efficacy of the drug.
- Clinical Trial Site
- The location where trial-related activities are actually conducted.
- Date of Commencement of a Clinical Trial
- For the purpose of the Clinical Trial Site Information Form, this is defined as the date when the clinical trial site will be ready to enrol patients in the clinical trial Footnote 1.
- Drug
- A drug [that is (i.e.) pharmaceuticals, biologics, gene therapies, blood products, vaccines and radiopharmaceuticals] for human use that is to be tested in a clinical trial.
- Good Clinical Practices
- Generally accepted clinical practices that are designed to ensure the protection of the rights, safety and well-being of clinical trial subjects and other persons, and the good clinical practices referred to in section C.05.010 of the Regulations.
- Import
- To import a drug into Canada for the purpose of sale in a clinical trial.
- Importer
- The sponsor or person designated by the sponsor who is responsible for the import of the drug into Canada for the purpose of sale in a clinical trial. Individual investigators at the clinical trial sites in Canada may serve as Canadian Importers.
- Informed Consent Form Footnote 2
- A document that describes:
- The risks and anticipated benefits to his or her health arising from participation in the clinical trial; and
- All other aspects of the clinical trial that are necessary for that person to make the decision to participate in the clinical trial.
- Institution/Investigator-initiated Clinical Trial
- A clinical trial that is initiated and conducted by an institution or an individual investigator. For such trials, the institution or investigator is considered to be the sponsor of the trial and must fulfill all the regulatory obligations of the sponsor as outlined in the Regulations.
- Investigator's Brochure
- In respect of a drug, a document containing the nonclinical and clinical data on the drug that are described in section C.05.005(e) of the Regulations.
- Phase I
- Clinical trials designed to determine the pharmacokinetics/pharmacological actions of the drug and the side effects associated with increasing doses. Drug interaction studies are usually considered as Phase I trials regardless of when they are conducted during drug development . Phase I trials are generally conducted in healthy volunteers , but may be conducted in patients when administration of the drug to healthy volunteers is not ethical.
- Phase II
- Clinical trials to evaluate the efficacy of the drug in patients with medical conditions to be treated, diagnosed or prevented, and to determine the side effects and risks associated with the drug. If a new indication for a marketed drug is to be investigated, then those clinical trials may generally be considered Phase II trials.
- Phase III
- Controlled or uncontrolled trials conducted after preliminary evidence suggesting efficacy of the drug has been demonstrated. These are intended to gather the additional information about the clinical efficacy and safety under the proposed conditions of use.
- Phase IV
- All studies performed within the approved indication after the drug has been approved by the regulator for the market . These studies are often important for optimizing the drug's use. They may be of any type but must have valid scientific objectives. Commonly conducted studies include safety studies and studies designed to support use under the approved indication (e.g., mortality and morbidity studies, or epidemiological studies).
- Protocol
- A document that describes the objectives, design, methodology, statistical considerations and organization of a clinical trial.
- Qualified Investigator
- The person responsible to the sponsor for the conduct of the clinical trial at the clinical trial site, who is entitled to provide health care under the laws of the province where that clinical trial site is located, and who is:
- in the case of a clinical trial respecting a drug to be used for dental purposes only, a physician or dentist and a member in good standing of a professional medical or dental association; and
- in any other case a physician and a member in good standing of a professional medical association.
- Research Ethics Board
- A body that is not affiliated with the sponsor, and
- the principal mandate of which is to approve the initiation of, and conduct periodic reviews of, biomedical research involving human subjects in order to ensure the protection of their rights, safety and well-being; and
- that has at least five members, that has a majority of members who are Canadian citizens or permanent residents under the Immigration Act, that is composed of both men and women and that includes at least:
- two members whose primary experience and expertise are in scientific discipline, who have broad experience in the methods and areas of research to be approved and one of whom is from a medical discipline or, if the clinical trial is in respect of a drug to be used for dental purposes only, is from a medical or dental discipline,
- one member knowledgeable in ethics,
- one member knowledgeable in Canadian laws relevant to the biomedical research to be approved,
- one member whose primary experience and expertise are in a non-scientific discipline, and
- one member who is from the community or is a representative of an organization interested in the areas of research to be approved and who is not affiliated with the sponsor or the site where the clinical trial is to be conducted.
- Sell
- Includes offer for sale, expose for sale, have in possession for sale and distribute, whether or not the distribution is made for consideration.
- Senior Executive Officer
- The senior executive officer is the most senior person with policy and operational decision making authority within the sponsor, or is an official who has this delegated authority in respect of the clinical trial. The SEO is responsible for providing an attestation with respect to the Clinical Trial Application/Amendment at the time of filing, as outlined in Appendix 3 of the Drug Submission Application Form (HC/SC 3011).
- Senior Medical or Scientific Officer
- A scientific or medical officer residing in Canada, representing the sponsor, who is responsible for providing an attestation with respect to the Clinical Trial Application/Amendment at the time of filing, as outlined in Appendix 3 of the Drug Submission Application Form (HC/SC 3011).
- Serious Adverse Drug Reaction
- An adverse drug reaction that requires in-patient hospitalization or prolongation of existing hospitalization, that causes congenital malformation, that results in persistent or significant disability or incapacity, that is life threatening or that results in death.
- Serious Unexpected Adverse Drug Reaction
- A serious adverse drug reaction that is not identified in nature, severity or frequency in the risk information set out in the investigator's brochure or on the label of the drug.
- Sponsor
- An individual, corporate body, institution or organization that conducts a clinical trial as per Division 5. The sponsor must comply with its obligations as set out in the Regulations (including C.05.010-C.05.015) in adhering to good clinical practices for the proper use of the drugs, drug labelling requirements, record keeping, submission of information, reporting of ADRs, and trial discontinuation reporting requirements.
- Study Completion
- Notwithstanding a suspension, cancellation or study closure of a clinical trial in Canada, in its entirety, a study is considered to be completed after the last subject globally completes the "end of study" visit as defined in the protocol. The "end of study visit" is the final visit for study-related tests and procedures, including the capture of any final potential study-related adverse events.
2.2 Pre-Clinical Trial Application (CTA) Consultation Meeting
Health Canada invites sponsors to request a pre-CTA consultation meeting. Such consultations may be particularly useful for new active substances or applications that will include complex issues that may be new to Health Canada.
The pre-CTA consultation meeting provides an opportunity for the sponsor to present relevant data, discuss concerns and issues regarding drug development. It also gives Health Canada an opportunity to provide guidance on the acceptability of the proposed trial(s). Sponsors may invite the qualified investigator(s) who will be involved in the proposed trial(s) in Canada to attend the meeting.
2.2.1 Request for a Pre-Clinical Trial Application (CTA) Consultation Meeting
Requests for a pre-CTA consultation meeting should be submitted in writing by the sponsor to the appropriate Directorate (refer to Appendix 1).
Requests should be submitted in the form of a cover letter proposing four dates and times suitable for a pre-CTA consultation meeting. The cover letter should be accompanied by the following information:
- A brief synopsis of the proposed study;
- A list of preliminary questions to be addressed by the Directorate during the meeting; and
- Sufficient information for Health Canada to assess the utility of the meeting and identify the appropriate staff necessary to discuss the proposed issues. This will assist in ensuring efficient use of Health Canada resources.
The Directorate will acknowledge the request for consultation in a timely manner. If the Directorate agrees with the request, the acknowledgement letter will confirm the pre-CTA consultation meeting date and indicate the number of copies of the pre-CTA information package to be provided 30 days before the confirmed meeting.
2.2.2 Pre-Clinical Trial Application (CTA) Information Package
The Information Package, which should be submitted in accordance with current electronic specifications (see Appendix 2), should contain:
- proposed agenda, any prepared slides including a finalized list of questions, and a complete list of attendees [it is recognized that the slides may change prior to the meeting.]
- a brief summary of all data including:
- a tabular listing of completed nonclinical and clinical studies,
- an outline of the observed toxicological manifestations and a discussion of their impact on the use of the drug in humans,
- an outline of the observed adverse events and a discussion of potential safety problems;
- a proposed global clinical plan for the current stage of drug development including regulatory status in other countries; [It is recognized that this plan is subject to change as new information becomes available.]
- details of the proposed clinical trials to be conducted in Canada, within the scope of the intended CTA, including:
- a statement of trial design,
- parameters, values, ranges or limits for indication(s) and clinical use(s), patient study population(s) and routes of administration,
- parameters, values, ranges or limits for dosage form(s), dosage regimen(s) and formulation(s),
- proposed procedures and/or criteria for patient monitoring, clinical efficacy and safety assessments, alternative treatments, premature patient discontinuation and other considerations, as appropriate;
- a summary of significant Quality (Chemistry and Manufacturing) aspects of the drug, if applicable;
- a summary of the method of manufacture for both drug substance and dosage form,
- relevant flow charts,
- a listing of quality control procedures and specifications,
- a summary of product characteristics, and
- a listing of all production site(s) - only for biologics and radiopharmaceuticals.
Should the pre-CTA package be found deficient, the sponsor may be requested to reschedule or postpone the meeting to allow the sponsor to assemble a more thorough package. Please note that the Directorate reserves the right to modify or truncate the proposed agenda as it sees fit to better achieve the stated goals of the meeting.
2.2.3 Pre-Clinical Trial Application (CTA) Consultation Meeting Record
The sponsor should prepare and send to the appropriate Directorate a written record of the discussions and conclusions of the consultation meeting within 14 days of the consultation date. All records of this consultation will be added to the Central Registry (CR) file for the drug.
A copy of the record of discussions and conclusions approved by all parties in attendance at the meeting should be included in the subsequent CTA.
2.3 Clinical Trial Applications (CTAs)
The sponsor must file a CTA prior to the initiation of the trial [C.05. 005]. CTAs are required for human clinical trials using drugs not authorized for sale in Canada, including clinical trials in Phases I through III of drug development and comparative bioavailability studies; as well as trials involving marketed drugs, where the proposed use of the drug is outside the parameters of the NOC or DIN , e.g., one or more of the following is different:
- Indication(s) and clinical use;
- Target patient populations(s);
- Route(s) of administration; or
- Dosage regimen(s).
Sponsors are not required to file a CTA for clinical trials involving marketed drugs where the investigation is to be conducted within the parameters of the approved NOC or DIN [C.05.006(2)]; these trials are referred to as Phase IV clinical trials.
Sponsors must conduct all clinical trials, including Phase IV trials, in accordance with Division 5, including the principles of GCPs, labelling requirements and obtaining REB approval.
Sponsors should register their clinical trials on one of two publicly accessible registries accepting international clinical trial information and recognized by the World Health Organisation (WHO): ClinicalTrials.gov and Current Controlled Trials International Standard Randomised Controlled Trials Number Register.
2.3.1 Filing a Clinical Trial Application (CTA)
CTAs should be sent directly to the appropriate review Directorate (refer to Appendix 1).
The outer label should be clearly identified with "Clinical Trial Application".
CTAs or CTA-As must be submitted to the appropriate lead Directorate / Bureau when they involve the use of:
- pharmaceuticals and biologics or radiopharmaceuticalsFootnote 3;
- a medical device and drug combination that is classified as a drug;Footnote 4 or
- a natural health product and a drugFootnote 5.
Authorization for the sale and importation of all investigational products to be used within a CTA or CTA-A must be obtained prior to the initiation of the clinical trial or implementation of the protocol amendment.
The lead Directorate / Bureau will be responsible for communicating the regulatory decision to the sponsor.
A separate Investigational Testing Application (ITA) and CTA must be filed and authorized before the trial can commence for protocols that involve a drug and the use of an unlicensed class II, III, or IV medical device that is not a combination product.
2.3.2 Clinical Trial Application (CTA) Format
The CTA is composed of three parts (modules) in accordance with the CTD format:
- Module 1 - contains administrative and clinical information about the proposed trial;
- Module 2 - contains Quality (Chemistry and Manufacturing) summaries about the drug product(s) to be used in the proposed trial; and
- Module 3 - contains additional supporting Quality information.
The CTA should be submitted on electronic media, accompanied by a hard copy cover letter, and be organized in accordance with the current electronic specifications: Guidance Document: Preparation of Drug Regulatory Activities in the "Non-eCTD Electronic-Only" Format.
Refer to Table 1 below for guidance on submission content, and to Appendix 2 (where applicable) for Guidance documents that may be useful in the preparation of the application.
Table 1: Contents of Submission Package in accordance with CTD Format
Module 1 Administrative and Product Information
- 1.0 Correspondence
- 1.0.1 Cover letter
- CTA/CTA-A: For CTA-As, a cover letter indicating the original CTA(s) and relevant CTA-As with file number and control number(s).
- CTA-A (Quality)For Biologics and Radiopharmaceuticals only: include a list of all proposed quality changes from the authorized application. Refer to section 2.4.2a.
- 1.0.5 Meeting Information
- CTA/CTA-A: Including e.g., a copy of the record of the discussions and conclusions of the pre-CTA consultation meeting or other relevant correspondence with Health Canada, if applicable.
- 1.0.1 Cover letter
- 1.1 Table of Contents
- CTA/CTA-A: A listing of the contents of Module 1 (Administrative / Clinical Information), Module 2 (Common Technical Document Summaries) and Module 3 (Quality), if applicable.
- 1.2 Administrative Information
- 1.2.1 Application Forms
- CTA/CTA-A: A completed and signed Drug Submission Application Form (HC/SC 3011) including Appendix 3, signed by the Senior Medical or Scientific Officer in Canada and the Senior Executive Officer. (Appendices 1 and 2 of the HC/SC 3011 Form should be completed and submitted if applicable). Please refer to Appendix 2 of this document for the relevant URL address.
For Institution/Investigator-initiated clinical trials, Appendix 3 of the Drug Submission Application Form (HC/SC 3011) may be signed by the appropriate department head in lieu of the Senior Executive Officer and the Qualified Investigator in lieu of the Senior Medical or Scientific Officer.
- CTA/CTA-A: A completed and signed Drug Submission Application Form (HC/SC 3011) including Appendix 3, signed by the Senior Medical or Scientific Officer in Canada and the Senior Executive Officer. (Appendices 1 and 2 of the HC/SC 3011 Form should be completed and submitted if applicable). Please refer to Appendix 2 of this document for the relevant URL address.
- 1.2.3 Certification and Attestation Forms
- CTA/CTA-A: Including the Summary of Additional Drugs Form (refer to Appendix 4).
- 1.2.5 Compliance and Site Information
- 1.2.5.1 Clinical Trial Site Information Form
- CTA: The Clinical Trial Site Information (CTSI) Form should be provided for each proposed clinical trial site, if known at the time of the application as per C.05.005(c). Health Canada recognizes that not all information required in the CTSI form may be available at the time of filing a CTA. Sponsors are reminded that even if this information is not available when filing the CTA, it is required prior to commencement of the trial as per C.05.006(1)(d). Please refer to section 2.7.3 for additional information.
If any changes are made to the CTSI Form (for example, change of qualified investigator) a revised CTSI Form should be submitted. - CTA-A (Clinical): An updated CTSI form should be provided for each site participating in the clinical trial to report that the amendment has been implemented. Health Canada recognizes that not all information required in the CTSI form may be available at the time of filing a CTA-A. Sponsors are reminded that even if this information is not available when filing the CTA-A, it is required prior to commencement of the trial as per C.05.008 (1)(c). Please see section 2.7.3 for additional information.
- CTA: The Clinical Trial Site Information (CTSI) Form should be provided for each proposed clinical trial site, if known at the time of the application as per C.05.005(c). Health Canada recognizes that not all information required in the CTSI form may be available at the time of filing a CTA. Sponsors are reminded that even if this information is not available when filing the CTA, it is required prior to commencement of the trial as per C.05.006(1)(d). Please refer to section 2.7.3 for additional information.
- 1.2.5.1 Clinical Trial Site Information Form
- 1.2.6 Authorization for Sharing Information
- CTA/CTA-A: Letters authorizing Health Canada to access related files (i.e.: a previously authorized CTA, Drug Master Files, Manufacturing Site Reference Files), if applicable. For example, a letter of access may be required to satisfy requirements for a CTA if a sponsor is utilizing a drug in a clinical trial that has not received a NOC and/or a DIN and the manufacturer of the drug does not wish to disclose confidential information about the drug to the clinical trial sponsor.
- Reference to a Drug Master File (DMF):
- A letter written by the holder of the DMF permitting Health Canada to reference information in the DMF in support of the sponsor's CTA should be submitted.
- The CTA sponsor should ensure that the supporting DMF (including submission of the letter of access and payment of related fees) has been submitted to and accepted by Health Canada prior to filing a CTA.
- Reference to an application previously submitted by a different sponsor and authorized by Health Canada:
- A letter written by the sponsor of the referenced application authorizing Health Canada to access the information in support of the CTA should be submitted.
- The referenced information should meet the regulatory requirements for CTAs.
- The letter of access should include the file number and control number(s) of the referenced submission(s).
- For Pharmaceuticals: Where chemistry and manufacturing information is referenced, sponsors are still required to complete the appropriate Quality Overall Summary (QOS) template (Module 2, [2.3]) including the introduction and any sections not covered by the letter of access.
- Reference to a Drug Master File (DMF):
- CTA/CTA-A: Letters authorizing Health Canada to access related files (i.e.: a previously authorized CTA, Drug Master Files, Manufacturing Site Reference Files), if applicable. For example, a letter of access may be required to satisfy requirements for a CTA if a sponsor is utilizing a drug in a clinical trial that has not received a NOC and/or a DIN and the manufacturer of the drug does not wish to disclose confidential information about the drug to the clinical trial sponsor.
- 1.2.7 International Information
- CTA/CTA-A (Clinical): Information regarding refusals by regulatory authorities outside Canada and foreign Research Ethics Boards, if applicable
- 1.2.9 Other Administrative Information
- CTA/CTA-A: This section is for any administrative information that does not have a designated location in the CTD format. This section should NOT contain any scientific information.
- 1.2.1 Application Forms
- 1.3 Product Information
- 1.3.4 Investigator's Brochure
- CTA: A copy of the current Investigator's Brochure (IB), supplemented as appropriate with up-to-date safety, non-clinical and clinical data.
The IB containing all information regarding the product to date should be prepared in accordance with the ICH E6, and should be reviewed at least annually and revised as necessary. If the IB has been updated relative to a version contained within a previously authorized CTA or CTA-A, a tabulated summary of the changes should be provided, including a rationale for each change that includes any omissions or deletions from previous versions if it has not been previously provided via a CTA-N. Sectional reports (section C.08.005.1(2) of the Regulations) should not be submitted unless requested. Please refer to section 2.8.4 for additional information.
For products marketed in Canada, a reference (including version date) to the most recent Canadian Product Monograph (PM) may be submitted if an updated IB is not available. - CTA-A (Clinical): If the CTA-A proposes to extend the duration of the treatment, an updated IB or equivalent information with supporting toxicological studies and clinical safety data to support the extension should be provided. The amendment to the IB may be included as an addendum.
- CTA-A (Quality): For Biologics and Radiopharmaceuticals only: a revised IB or an Addendum to the IB describing any new Quality (Chemistry and Manufacturing) information, including supporting data as required, if applicable.
- CTA: A copy of the current Investigator's Brochure (IB), supplemented as appropriate with up-to-date safety, non-clinical and clinical data.
- 1.3.4 Investigator's Brochure
- 1.4 Health Canada Summaries
- 1.4.1 Protocol Safety and Efficacy Assessment Template - Clinical Trial Application (PSEAT-CTA)
- CTA: A Protocol Synopsis in the format PSEAT-CTA.
- 1.4.1 Protocol Safety and Efficacy Assessment Template - Clinical Trial Application (PSEAT-CTA)
- 1.7 Clinical Trial Information
- 1.7.1 Protocol
- CTA: A copy of the final proposed protocol(s), including version number.
- CTA-A (Clinical): A copy of the amended or working protocol with a clear description of the changes that are being proposed (that is, original wording vs. revised wording), a rationale for each proposed change, and a copy of the most recently authorized protocol, including version number.
The changes may be listed in a separate document or an annotated version of the protocol. Cross-referencing is not acceptable.
- 1.7.2 Informed Consent Forms
- CTA: A copy of the Informed Consent Forms (ICFs) to be used in conjunction with the clinical trial, including a statement regarding the risks and anticipated benefits to the clinical trial subjects as a result of their participation in the clinical trial. ICFs to be used in conjunction with the clinical trial should be prepared in accordance with applicable laws governing consent. The ICH E6 and the Tri-Council Policy Statement (TCPS) provides standards for the ICF.
- CTA-A (Clinical): The revised ICFs must be submitted if the changes to the study protocol(s) or other supporting documentation (nonclinical study results, adverse events, revisions to the IB) affect the information in the ICF. The ICF with changes clearly indicated (annotated) should be provided.
- 1.7.3 Canadian Research Ethics Board (REB) Refusals
- CTA/CTA-A (Clinical): The name, address and telephone number and, if applicable, the fax number and electronic mail address of any REB that has previously refused to approve the clinical trial protocol or amendment, its reasons for doing so and the date on which the refusal was given, if known at the time of submitting the application as per C.05.008(1)(c). Please refer to section 2.7.1 for additional information.
- 1.7.4 Information on Prior-related Applications
- CTA/CTA-A: A list of ongoing clinical trials in Canada for which authorization has been granted by Health Canada, if applicab
- 1.7.1 Protocol
Module 2 Common Technical Document Summaries
This module contains Quality (Chemistry and Manufacturing) Information only. This section does not apply if the drug product to be used in the clinical trial has received a NOC and/or DIN and has not been modified.
If the Quality information was previously submitted to, and authorized by Health Canada and has not changed, re-submission of the applicable Quality Summary may not be required. However, sponsors should refer to the control number of the prior application.
The Common Technical Document Summaries Module should include:
CTA-A (Quality): An applicable updated Quality Overall Summary (QOS) or Quality Information Summary (QIS) containing only the revised sections. The rationale for each proposed change should be submitted and the revised information should be clearly identified. Alternatively, the changes may be listed in a separate document or in a marked up annotated version of the QOS/QIS-R/QIS-PER, as applicable.
- 2.1 Table of Contents
- CTA/CTA-A (Quality): A listing of the contents of Modules 2 and 3, if applicable.
- 2.3 Quality Overall Summary (QOS)
- CTA
- For Pharmaceuticals: a QOS is required [QOS-CE (CTA - Phase I), QOS-CE (CTA - Phase II), QOS-CE (CTA - Phase III)]
For placebo-controlled studies, a qualitative list of the ingredients in the placebo should be submitted. - For Biologics and Radiopharmaceuticals:
There are four QOS Guidance documents to be used as direction for the completion of the quality section for biologic drug submissions (refer to Appendix 2 for links) and two QIS (Quality Information Summary) templates for radiopharmaceutical drug submission applications [QIS-R and QIS-PER 9 (not yet posted externally as of January 2011)]. The applicant should submit a completed QOS/QIS with, as a minimum, those subsections or parts which have a check mark () beside the guidance or heading, including the facility information. Note that these guidances were not written specifically for CTAs and may not necessarily apply to the same extent. It is understandable that depending upon the stage of drug development, a limited amount of information may be available for a CTA; in which case, the sponsor should provide whatever data are available at that time. Sponsors should also refer to the applicable Health Canada quality Guidance documents and updated notices for additional information.
For placebo-controlled studies, information on the placebo is also required including a description of the manufacturing process, a qualitative and quantitative list of ingredients, specifications, batches, stability and facility information.
- For Pharmaceuticals: a QOS is required [QOS-CE (CTA - Phase I), QOS-CE (CTA - Phase II), QOS-CE (CTA - Phase III)]
- CTA
Module 3 Quality (if submitted)
- 3.1 Table of Contents of Module 3
- CTA/CTA-A (Quality): A listing of the contents of Module 3.
- 3.2 Body of Data
- CTA/CTA-A (Quality): Where there is additional supporting quality information provided in the QOS-CE (Module 2), this information should be provided separately in the appropriate Module 3 section and cross-referenced in the applicable QOS/QIS. Sponsors should refer to the applicable Health Canada quality Guidance documents for additional information.
For Biologics and Radiopharmaceuticals: For a product early in development, submission of Module 3 is not always necessary if sufficient information is provided in the QOS/QIS-R/QIS-PER, as appropriate.
- CTA/CTA-A (Quality): Where there is additional supporting quality information provided in the QOS-CE (Module 2), this information should be provided separately in the appropriate Module 3 section and cross-referenced in the applicable QOS/QIS. Sponsors should refer to the applicable Health Canada quality Guidance documents for additional information.
- 3.3 Literature References
- CTA/CTA-A (Quality): Literature references related to quality information should be provided here if applicable.
2.3.3 Comparative Bioavailability Trial Application Requirements (Seven Day Administrative Review)
Health Canada's administrative seven (7) day review target applies to applications involving comparative bioavailability studies for pharmaceuticals only where:
- The studies are single-dose studies to be performed on healthy adult volunteers;
- The reference drug product is marketed in Canada, United States, European Union, Australia, Switzerland or Japan; and
- The maximum dose of the study drug does not exceed that specified in the labelling of the reference drug product.
This section does not apply to biologics, radiopharmaceuticals and cellular therapies, which includes Phase I trials using somatic cell therapies, xenografts, gene therapies, prophylactic vaccines or reproductive and genetic technologies. Additionally, this section does not apply to other comparative bioavailability studies, such as those conducted during drug development of new active substances to assess the impact of changes to dosage forms or manufacturing processes, or studies comparing different routes of administration. For those types of studies please refer to section 2.3.2 for filing requirements and section 2.5 for review process and timelines.
CTAs for comparative bioavailability studies should be filed directly to the Therapeutic Products Directorate, addressed to the attention of the Director. The outer label of the shipping carton should be clearly identified with "Clinical Trial Application for Bioavailability Studies". In general, the CTA filing requirements (section 2.3.2) also apply to the comparative bioavailability studies that meet the criteria provided above, with some exceptions as follows:
- The cover letter to the application should include a rationale for the study;
- The current labelling or PM/Prescribing Information for the reference product in lieu of the IB; and
- A completed Quality Overall Summary - Chemical Entities (Clinical Trial Application - Bioavailability Studies) (QOS-CE (CTA-BA)) template, as well as any additional quality information as outlined in the template.
CTA-A and CTA-Notification (CTA-N) filing requirements (refer to sections 2.4 and 2.6, respectively) also apply to comparative bioavailability studies.
2.4 Clinical Trial Application-Amendments (CTA-As)
CTA-As are applications in which a sponsor proposes information to support changes to a previously authorized application [C.05.008]. CTA-As are required for changes to clinical trial drug supplies that affect the quality or safety of the drug, changes to an authorized protocol that alter the risk to clinical trial subjects, or both.
CTA-As must be authorized by Health Canada prior to implementation of the changes [C.05.008]. However, if the sponsor is required to immediately make one or more of the amendments referred to in subsection (2) of C.05.008 because the clinical trial or the use of the drug for the purposes of the clinical trial endangers the health of clinical trial subjects or other persons, the sponsor may immediately make the amendment without prior review by Health Canada. Sponsors must notify Health Canada of this change, provide the relevant rationale in support of the immediate implementation and file a CTA-A that clearly identifies the change and the rationale for immediate implementation of the change within 15 days after the date of implementation of the amendment [C.05.008(4)].
Amendments submitted when the CTA is under review will not be accepted. Where a sponsor wishes to make changes to the CTA under review, the sponsor should withdraw the active CTA and submit the amendment as a new CTA.
2.4.1 Clinical Trial Application-Amendment (CTA-A): Clinical
Sponsors are required to file CTA-As for changes to the protocol made after the original CTA that will impact on the safety of the subjects or will affect the analysis and the interpretation of the safety and efficacy of the drug(s) under investigation. As per section C.05.008(2), a CTA-A must be filed when the proposed amendments to the protocol:
- Affect the selection, assessment, or dismissal of a clinical trial subject;
- Affect the evaluation of the clinical efficacy of the drug;
- Alter the risk to the health of a clinical trial subject;
- Affect the safety evaluation of the drug; or
- Extend the duration of the treatment.
Examples of protocol changes that require a CTA-A are provided below to aid in determining whether a CTA-A should be filed. These examples are not all inclusive. When in doubt of whether a CTA-A is warranted sponsors should contact the corresponding Directorate.
Examples include, but are not limited to:
- Criteria, tests or procedures required to select or dismiss a clinical trial subject. These include changes to eligibility criteria, tests or procedures for selecting the study population, as well as tests, procedures, or criteria for dismissing clinical trial subjects prematurely or at the end of the trial;
- Criteria, tests or procedures required for the ongoing assessment of clinical trial subjects, including assessment of safety, or evaluation of safety and efficacy. This includes protocol changes as a result of serious unexpected ADRs;
- Study design, study population, duration of use, objectives, or hypotheses, including adding or discontinuing a study arm that was not included as a provision in the original CTA protocol;
- Changes in the primary efficacy endpoint(s), important secondary efficacy endpoints [for example (e.g.), those that could be used in support of a marketing application], safety endpoints, sample size estimation, or addition of interim analyses that will affect the analysis and interpretation of the study results;
- Dose level, dosage schedule, or treatment duration;
- Changes to the post-treatment follow-up period that may affect the safety evaluation of the drug.
- Adding or removing a concomitant medication, which may impact on the analysis of efficacy or increase the risk to clinical trial subjects;
- Criteria for expedited reporting of serious, unexpected adverse drug reactions;
- Increases in blood volume, changes in procedures, enrolling additional subjects in PK studies or confirmatory testing in PK studies that were not specified in the original CTA protocol; and/or
- Aspects of the conduct of the study that may increase the risk to the health of clinical trial subjects.
Protocol changes should be reflected in a revised ICF, as applicable. Additionally, new information related to the safety of the drug may affect a subject's decision to participate in the trial, and should be added to the risks section of the ICF. An updated copy of the ICF should be included in the CTA-A, as applicable, with changes clearly indicated (annotated). Refer to section 2.4.3 for additional information on filing a Clinical CTA-A.
Protocol changes that extend the duration of the clinical trial pertain to extensions in the treatment period of individual study subjects. All protocol changes that involve an extension in treatment duration or treatment period require filing of a CTA-A; such CTA-As must be accompanied by an IB or equivalent information to support the extension in treatment duration. Changes in the projected duration of the entire trial, not impacting individual patients' duration of treatment, are normally not considered to require a CTA-A.
2.4.2 Clinical Trial Application-Amendments (CTA-A) and Clinical Trial Application- Notification (CTA-N): Quality (Chemistry and Manufacturing)
Sponsors must file a CTA-A or CTA-N to a previously authorized application when changes that may affect the quality or safety of the clinical trial drug supplies are proposed. Changes to the Quality summary subsections of Module 2 and Module 3 (if applicable) including, but not limited to those listed in Tables 2 to 5 below, warrant the filing of a CTA-A or a CTA-N.
a) For Biologics and Radiopharmaceuticals
A list of all proposed quality changes from the authorized application should be provided in the cover letter.
It should be noted that for Biologics and Radiopharmaceuticals, differences in manufacturing strategies can lead to the production of a novel drug product requiring both non clinical and clinical data to support its use and are considered beyond the scope of an authorized CTA. In such cases, a new CTA is required. Examples of differences in manufacturing strategies include, but are not limited to:
- Change in the source of drug substance (e.g., from a fermentation process to transgenic milk);
- Change in the host cells used to express the same coding sequence;
- Change in the strain of virus used in manufacturing a vaccine;
- Change in the strain of oncolytic virus used in cancer treatment;
- Change in the animal source of an immune globulin (e.g., from rabbit to sheep);
- Change in the source of a radionuclide (e.g., from nuclear reactor to cyclotron or linear accelerator) for labelling kits;
- Change in the source of the parent radionuclide (e.g., from nuclear reactor to cyclotron or linear accelerator) used in a generator;
- Change in the design, structure and operation of a radionuclidic generator.
For additional guidance regarding the classification of a quality change, sponsors are encouraged to consult with BGTD.
For a product commercially available and used in clinical trials for which a quality change has been made according to the Post-NOC Changes Guidance document, supporting data are not required in support of the same change affecting the clinical product. The change can be notified to the BGTD with cross-reference to the approved submission filed for the commercial product. In the situation where a change made to the commercial product has not yet been approved and is affecting the clinical material, a CTA-A or a CTA-N must be submitted according to the tables below. For Level 3 changes made to a biologic/radiopharmaceutical, a CTA-N is not required for the clinical product.
| Type of Change | Submission Type | |
|---|---|---|
| 1. Replacement or addition of a manufacturing site involving: | a. production of the starting material, intermediate, or drug substance | Amendment |
| b. testing [for example (e.g.), release, stability] | Notification | |
| 2. Change in the manufacturing process for the drug substance intermediate, involving: | a. the fermentation process [for example (e.g.), scale-up, new bioreactor technology, use of new raw materials of biological origin]; or change in the route of synthesis of the radiopharmaceutical drug substance or critical componentTable 1 footnote 1 | Amendment |
| b. the purification process (e.g., addition/removal/replacement of a purification step) | Amendment | |
| 3. Change in the specifications for the drug substance involving: | a. deletion or replacement of a test, relaxation of an acceptance criterion or addition of a test for a new impurity | Amendment |
| b. addition of a test (other than a test for new impurity) or tightening of an acceptance criterion | Notification | |
| 4. Change in the primary container closure system(s) for the storage and shipment of the drug substance provided the proposed container closure system is at least equivalent to the approved container closure system with respect to its relevant properties, and the change does not concern a sterile drug substance | Notification | |
| 5. Change in the shelf life for the drug substance, involving: | a. (i) Extension if the approved shelf life is less than or equal to 18 months | Amendment |
| a. (ii) Extension if the approved shelf life is more than 18 months | Notification | |
| b. Reduction (due to stability concerns) | Amendment | |
| Type of Change | Submission Type | |
|---|---|---|
| 1. Replacement or addition of a drug product manufacturing site involving: | a. production of a drug product (including primary packaging) | Amendment |
| b. secondary packaging | Notification | |
| c. testing [for example (e.g.), release, stability] | Notification | |
| 2. Change in the drug product manufacturing process (e.g., scale-up, changes to the formulation process); change from manual synthesis of positron-emitting radiopharmaceutical to use of Automatic Synthesis Unit (ASU) or change in type of ASU | Amendment | |
| 3. Deletion of a drug product manufacturer / manufacturing site, primary or secondary packaging site or testing site | Notification | |
| 4. Change in the specifications for the drug product, involving: | a. deletion or replacement of a test, relaxation of an acceptance criterion or addition of a test for a new impurity | Amendment |
| b. addition of a test (other than a test for new impurity) or tightening of an acceptance criterion | Notification | |
| 5. Change in the shelf life for the drug product, involving: | a. (i) Extension - if the approved shelf life is less than or equal to 18 months | Amendment |
| a. (ii) Extension - if the approved shelf life is more than 18 months | Notification | |
| b. Reduction (due to stability concerns) | Amendment | |
| 6. Change in the storage conditions for the drug product | Amendment | |
| 7. Changes in final product dosage form (e.g., liquid to lyophilized formulation) | Amendment | |
| 8. Changes in final product strength | Amendment | |
| 9. Change in diluent, involving replacement or addition of a diluent for a lyophilized powder or concentrated solution by a diluent which is commercially available in Canada, is water for injection (WFI) or a salt solution, and after reconstitution, there is no change in the drug product specifications outside of the approved ranges. | Notification | |
| 10. Change in radiolytic protective agent or antioxidant | Amendment | |
For a product commercially available and used in clinical trials for which a quality change has been made according to the Post-NOC Changes Guidance, supporting data are not required in support of the same change affecting the clinical product. The change can be notified to the TPD with cross-reference to the approved submission filed for the commercial product. In the situation where a change made to the commercial product has not yet been approved and is affecting the clinical material, a CTA-A or a CTA-N must be submitted according to the table below.
| Type of Change | Submission Type | |
|---|---|---|
| 1. Replacement or addition of a manufacturing site involving: | a. production of drug substance | Amendment |
| b. testing [for example (e.g.), release, stability] | Notification | |
| 2. Change in the manufacturing process for the drug substance intermediate or starting material (e.g., reaction conditions, solvents, catalysts, synthetic routes, reagents, etc.) | Amendment | |
| 3. Change in the batch size for the drug substance (no impact on quality) | Notification | |
| 4. Change in the specification for the drug substance involving test and acceptance criteria: | a. Deletion or replacement of a test, relaxation of an acceptance criterion, or addition of a test for a new impurity | Amendment |
| b. addition of a test (other than a test for a new impurity) or tightening of an acceptance criterion | Notification | |
| 5. Change in the re-test period (or shelf life) for the drug substance, involving: | a. Extension | Notification |
| b. Reduction (due to stability concerns) | Amendment | |
| Type of Change | Submission Type | |
|---|---|---|
| 1. Addition of a dosage form or strength | Amendment | |
| 2. Change in the composition of a dosage form | Amendment | |
| 3. Qualitative or quantitative addition, deletion or replacement of a colour or flavour with no negative impact on stability | Notification | |
| 4. Change in diluent, involving replacement or addition of a diluent for a lyophilized powder or concentrated solution | Amendment | |
| 5. Replacement or addition of a drug product manufacturer / manufacturing site involving: | a. Production of an immediate release drug product (tablet, capsule, liquids, semi-solids) within the same Manufacturer | Notification |
| b. Production of an immediate release drug product (tablet, capsule, liquids, semi-solids) to a new Manufacturer | Amendment | |
| c. Production of a modified release product | Amendment | |
| d. Production of a sterile drug product | Amendment | |
| e. Primary packaging (non-sterile products) | Notification | |
| f. Testing [for example (e.g.), release, stability] | Notification | |
| 6. Change in the drug product manufacturing process | Amendment | |
| 7. Change in the specification for the drug product tests and acceptance criteria, involving: | a. Deletion or replacement of a test, relaxation of an acceptance criterion, or addition of a test for a new impurity | Amendment |
| b. addition of a test (other than a test for a new impurity) or tightening of an acceptance criterion | Notification | |
| 8. Change in the shelf life for the drug product, involving: | a. Extension | Notification |
| b. Reduction (due to stability concerns) | Amendment | |
| 9. Change in the storage conditions for the drug product | Amendment | |
2.4.3 Filing a Clinical Trial Application-Amendment (CTA-A)
CTA-As should be filed directly to the appropriate Directorate (Appendix 1). The outer label should be clearly labelled with "Clinical Trial Application - Amendment".
For joint reviews, refer to section 2.3.1.1.
2.4.4 Clinical Trial Application-Amendment (CTA-A) Format
Similar to CTAs, CTA-As should be organized and numbered as per the CTD format.
CTA-As should be submitted on electronic media, accompanied by a hard copy cover letter, and be organized in accordance with the current electronic specifications; Guidance Document: Preparation of Drug Regulatory Activities in the "Non-eCTD Electronic-Only" Format.
2.4.4.1 Clinical Amendments
Please refer to section 2.3.2 CTA Format for guidance in completing filing requirements for Clinical CTA-As; subsection 1.4.1 is not applicable.
2.4.4.2 Quality (Chemistry and Manufacturing) Amendments
Please refer to section 2.3.2 CTA Format for guidance in completing filing requirements; subsections 1.2.5.1, 1.2.7, 1.4.1, and 1.7.2 are not applicable.
2.5 Clinical Trial Application (CTA) and Clinical Trial Application-Amendments (CTA-A) Review Process
Health Canada reviews the documents submitted in CTAs and CTA-As to assess the quality of the products and determine that the use of the drug for the purposes of the clinical trial does not endanger the health of clinical trial subjects or other persons, the clinical trial is not contrary to the best interests of a clinical trial subject, and the objectives of the clinical trial may be achieved [C.005.006(1)(b)(ii)]. All CTAs, including those for comparative bioavailability studies, are subject to the 30 day default period from the date of receipt of the completed application as per C.05.005 or C.05.008.
Comparative bioavailability studies that meet the Comparative Bioavailability Trial Application Requirements (section 2.3.3) are targeted to be reviewed within 7 days. Sponsors are reminded that this expedited review process is an administrative target.
An acknowledgement letter will be issued to indicate the start of the review period and to indicate that the Minister is in receipt of a complete application.
This acknowledgement letter will also advise sponsors that if the CTA is authorized, and involves a trial in patients (phase I, II, or III), Health Canada will publish the following information about the clinical trial in Health Canada's publicly accessible database soon after an NOL is issued:
- protocol number
- protocol title
- drug name
- medical condition
- study population
- authorization date
- sponsor name
- Health Canada control number, and
- the start and end date of the clinical trial, if known.
All CTAs and CTA-As will be screened for completeness and if deficiencies are identified at screening, these will be addressed by a Request for Clarification or a Screening Rejection Letter. If the application is considered complete, an acknowledgement letter will be issued to indicate that the 30-day default period commenced on the date of receipt in Health Canada.
2.5.1.1 Requests for Clarification during screening
MIL-HDBK-61A: Configuration Control
< Previous | Contents | Next >
6.2 Engineering Change Proposal
An Engineering Change Proposal (ECP) is the management tool used to propose a configuration change to a CI and its Government-baselined performance requirements and configuration documentation during acquisition (and during post-acquisition if the Government is the CDCA for the configuration documentation).
The following paragraphs define uniform concepts and principles by which the processing of ECPs is conducted. These standard ground rules are necessary to assure that there is a consistency and orderly process that can be expeditiously accomplished by all parties.
The concepts in this section apply to class I ECPs, except where specifically identified as applicable to class II ECPs.
6.2.1.1 ECP Initiation.
The initiation of an ECP begins at the government's request unless for one or more of the reasons cited in paragraph b. below. Since most ECPs occur in a sole source environment, the initiation of an ECP should be a well-planned and coordinated effort between the government and contractor. A clear mutual understanding of the ECP objective, technical scope and the Government's performance, cost and schedule constraints shortens the lead-time for ECP preparation. It also results in a complete and comprehensive proposal to facilitate timely and effective implementation. As with most processes, the three C's: Communication, Cooperation and Coordination are the keys to assuring successful change processing.
The "ECP Management Guide," [Detail: Appendix D] has been developed to assist both the Government and contractor during the request, preparation, approval and implementing phases of an ECP. It provides checklists to aid in the timely identification and coordination of essential technical information required for decision making in all three stages of the ECP process. It also fosters the integrated product and process team concept.
a. Solicited ECPs.
Whenever the government identifies a need or requirement to change a CI and its configuration documentation a Class I ECP is formally requested from the contractor. A request for an ECP is coordinated with the applicable government Contracting Officer prior to being released to the contractor. [Refer to: Check List (A) of Appendix D]
b. Unsolicited ECPs.
As a general rule, unsolicited Class I ECPs are discouraged. However, at the discretion of the procuring activity, a preliminary ECP may be submitted to allow evaluation of the desirability of expending resources to fully document a proposed change. Changes that impact the following areas are instances where unsolicited ECPs may be justified:
Safety
Compatibility.
Correction of Defects.
Survivability.
Security.
Product improvement(s) that may significantly reduce life cycle costs, including Value Engineering Change Proposals (VECP) consistent with the DFAR Value Engineering clause of the applicable contract
Technology improvements
6.2.1.2 ECP Preparation and Submittal.
Formal and preliminary ECPs are prepared and submitted to the Government in accordance with the configuration management requirements of the applicable contract SOW and associated Contract Data Requirements List (CDRL), DD Form 1423 citing the latest approved Data Item Description (DID) for submittal of ECP data. The contract CDRL should provide information on submittal and distribution of ECPs for Government review and processing.
The contractor (ECP Originator) should notify the Government immediately by electronic message (e.g. E-mail, Fax) when the need for an emergency or urgent priority ECP is determined. Follow-up to a message ECP should be in the form of a formal ECP submittal, within 30 days. However when this is impracticable, a preliminary ECP may be used as an interim measure. Both the preliminary ECP (if used) and the final ECP resulting from a message ECP would be identified as revisions of the initial message ECP. [Detail: Activity Guide: Tables 6-3 and 6-4]
a. Automated Processing of ECPs.
If the Government has established a Government Configuration Management Automated Information System (CM AIS) the contract data requirement for ECPs should request either the digital submittal of ECP data, population of the DoD data base directly by the contractor, or access to the ECP via the world wide web. All ECP fields of information will be defined in the EIA Standard 836 data dictionary and its related XML ECP Business Object. To use MEARS as a standalone system, software must be provided to the contractor.
b. ECP Content by Program Life Cycle Phase.
Pertinent data fields of information (ECP data elements) that are to be provided by an ECP should be identified as described in the data item description and EIA-836. Only data fields that are populated need be provided with the ECP. Using the XML data field tags will enable Government and the various commercial configuration management information systems to store and coherently display the ECP data. A significant advantage of using electronic commerce over paper forms is that each topic may be addressed in its entirety without having to meet paper form block limitations. Obviously those key data fields that identify and describe the change are mandatory in any ECP. Common sense and the current context and environment of the program for which the ECP is being submitted dictate the fields to be populated. The typical content of an ECP may vary considerably during the CI's life cycle, and because DoD Directive 5000.1 gives Government Program Managers latitude in identifying the phases that they will employ, no two programs will necessarily be the same. The content guidance provided herein [Detail: Activity Guide: Table 6-6] reflects the general variability of ECP content that can be expected.
6.2.1.3 ECP Supporting Data.
Supporting data should include, where necessary, supplementary information to support the change description and justify the need for change. Test data, analyses and other technical documentation providing supporting rationale for assertions made in the ECP, and upon which the configuration control authority can base its acceptance of the proposed change, can be included to the extent that the originator feels is necessary. In many cases, the proposed change or its justification will be easier to understand if "marked-up" copies or draft revisions of the TDP element (such as a "redlined" copy of a portion of a specification or an interface drawing, or a draft table providing new values to be included in a data base) are also provided as a part of the ECP package.
6.2.1.4 Review and Dispositioning ECPs.
In order to facilitate dispositioning ECPs affecting documents for which the Government is CDCA, contracts should identify the government representative(s) responsible for dispositioning both Class I and Class II ECPs. Where the Government is an Application Activity (AA), or in a performance based acquisition, where the Government is not CDCA for the design documentation, contracts should clearly specify Government and contractor responsibilities for Class I ECPs and RFDs affecting Government baselined performance specifications. This can be accomplished by incorporating a special configuration control clause in the contract similar to the example in the box below. Guides for the dispositioning of Class I and Class II changes are provided in 6.2.2. [Detail: Activity Guide: Table 6-7] Key aspects of this process are highlighted, as follows:
Example:

a. Dispositioning Class I ECPs.
Class I ECPs must be dispositioned (approved or disapproved) for implementation by a properly constituted Government Configuration Control Board (CCB).[See 6.1.1.3.a.] After the CCB direction is issued, it is important to proceed expeditiously with the "definitization" process (obtaining a pricing proposal, auditing, fact finding, and negotiating the final price) for this change and issuing a supplemental agreement. Until the contract modification is received and bi-laterally agreed to by the Government and the contractor, the contractor is not authorized to proceed with the implementation of the proposed change.
The contractual approval or disapproval of an ECP should not be confused with the acceptance and approval of the ECP as a data deliverable. Approval of the ECP data delivery required by CDRL/DD Form 1423 signifies only that the ECP satisfies the requirements of the ECP DID and is considered acceptable for government processing. Acceptance of the data deliverable does not signify "technical approval" of the change proposed by the ECP and should not be interpreted as authorizing the performing activity(s) to proceed with the work proposed by the ECP.
All ECPs should be dispositioned by the Government as expeditiously as possible. The ECP indicates a date by which contractual authorization is required. This date should normally be proposed by the contractor to allow sufficient processing time by the Government. In some cases, expedited processing may be necessary in order to minimize the cost of the change or to enable it to be incorporated in time to satisfy an operational need. Since certain critical factors (such as safety or national defense preparedness) may be involved, it is important that the Government proceed with all due speed, but it is also important to ensure that proper priorities and need dates are being specified.
Because there is considerable urgency involved in effecting the changes proposed in urgent and emergency ECPs, the contractor normally specifies an authorization suspense date that is very close to the submittal date (e.g. 48 hours to make the technical decision on an emergency ECP and 30 calendar days to make the decision on an urgent ECP). [Detail: Activity Guide: Table 6-5.]
When the urgent or emergency priority is properly used, the contractor must be authorized to proceed with implementing the change as quickly as possible. Under these circumstances, it is often necessary to utilize a unilateral change order to the contract (or contracting officer letter) to provide official authorization to proceed. If the change order is to be used, a "not-to-exceed" price quotation (a "not-less-than" price for cost reduction ECPs) would be required to set a limitation on the price impact of the change activities to be accomplished. After the change order is issued, it is important to proceed as expeditiously as possible with the normal "definitization" process to minimize the risk of related price increase (or to maximize the related savings) resulting from the change.
VECPs are subject to essentially the same CCB process as other ECPs. Under the FAR clause, the Government is entitled to reimbursement of expenses incurred in processing an approved VECP before any cost savings are shared out to the contractor. Therefore, the tasking activity must develop auditable government cost information so that the complete monetary impact of the VECP can be evaluated. Any delays in VECP processing will typically reduce the savings benefit.
b. Dispositioning Class II ECPs.
Unless otherwise specified by contract (e.g., as part of the Single Process Initiative), the government administrative contracting officer or plant representative serves as the dispositioning authority for Class II ECPs.. The default action required on Class II changes is concurrence/nonconcurrence in classification only, unless the contract requires approval/disapproval. Government concurrence in Class II ECP classification normally allows the contractor to incorporate the change in the applicable CI and update its configuration documentation without any further government action or authorization being required. A nonconcurrence in classification will normally result in the Class II ECP being canceled or reclassified to a Class I ECP.
The government should require approval/disapproval of class II ECPS only when the Government is the CDCA for the original drawings, or data files, and compliance with the specific detailed design is a requirement of the contract. If there is a government ACO or plant representative available, the Government tasking activity may elect to have the ACO or representative review the proposed class II changes for concurrence in classification before they are submitted to the government tasking/procuring activity (that is the CDCA) for approval [Details: Activity Guide: Table 6-7]
6.2.1.5 Implementing Class I ECPs.
When ECPs are approved, change implementation to a CI being produced under contract is usually a straightforward contractual incorporation of the ECP as approved by the government CCB. CCB approval action is not to be considered authority for the contractor or tasking activity to proceed with the change.
A CCB directive must be prepared, published and distributed. The CCB directive is identified by the CCB identifier and the change identifier. The date of the CCB directive and disposition are recorded. Distribution should be limited to those parties required to take action to implement the change
If implementation of the approved change is the responsibility of the contractor under the terms of a contract, the CCB approval action directs the procurement contracting officer to initiate instructions to the contractor
If Contractor-initiated change proposals are involved, the receipt of a formal contract change for example, Standard Form 30, "Amendment of Solicitation/Modification of contract" or PCO letter (pending receipt of an amendment) shall constitute sole authority for the contractor to proceed.
If the initiator is government activity acting in the capacity of a contractor, the receipt of the directive/order (including funding authorizations) shall constitute sole authority to proceed with the change.
Change implementation to a CI in the inventory or operational forces will normally require the coordination of additional requirements of an implementing CCB directive (or tasking order).
Necessary instructions and funding authorizations must be issued for the scheduled implementation of the changee
Change accomplishment reporting is directed. [Details: Activity Guide: Table 6-8]
The incorporation of approved changes should be planned so that optimum acquisition, production, tests, evaluation and operational advantages can be derived from the modified configuration. The change is effectively coordinated to ensure that the earliest possible availability and support of the CI is provided with minimum disruptive effect on planned operating cycles.
Changes shall be incorporated only after the Contract modification/PCO letter or implementing directive/order is published and logistic support is available, unless safety or critical mission requirements dictate otherwise. Unofficial or preliminary technical documents shall not be used as authority to incorporate changes.
The implementation of approved changes to a CI must always include the proposed incorporation of new and revised technical documentation. Provisions for change documentation should always be addressed by the change proposal, contract modification and/or CCB implementing directive/order. Change documentation may include such types of data as specifications, drawings, provisioning documentation, technical manuals, diagrams, sketches, parts lists, master configuration lists, computer program documentation, and test and evaluation procedures. Requirements for such change documentation may vary depending on the life-cycle phase, type and complexity of each CI and the change/modification. However, the documentation prepared for any change will normally include the following three categories:
The documentation package (including the CCB implementing directive/order) forwarded to the change installing activities to install the change.
The documentation required by the technical, training, maintenance, and supply management organizations to properly control and support the change.
The documentation (e.g., technical manuals) required by the user activities to properly operate and maintain the CI after the change is installed.
The following ECP Activity Guides provide information concerning change classification, the justification for Class I ECPs, the types of ECPs, ECP priorities, ECP content, and the ECP dispositioning actions that may apply. ECPs are prepared and submitted to the government in accordance with the configuration requirements of the applicable contract SOW and CDRL/DD Form 1423. If the Government has established a CM AIS, the data requirement for ECPs should request digital submittal of ECP data, population of the DoD database directly by the contractor.
Table 6-2. Activity Guide: Change Classification


Table 6-3. Activity Guide: ECP Justification Codes
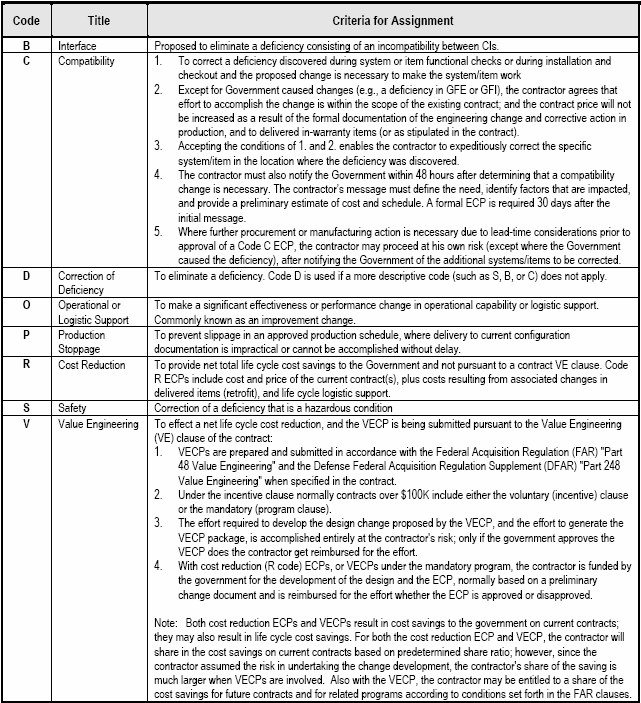
Table 6-4. Activity Guide: Class I ECP Types and Their Function
All types of Class I ECPS may be submitted to the Government electronically, the type categorization relates not to format but to give a quick indication of the intent of the ECP
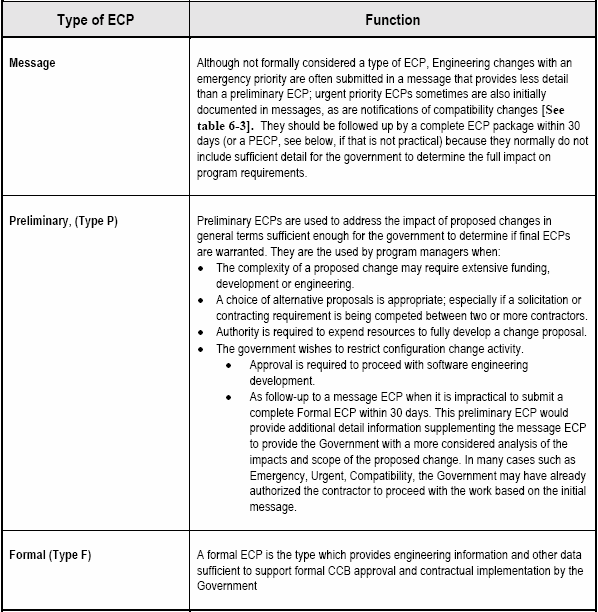
Table 6-5. Activity Guide: ECP Priorities
One of the following priorities shall be assigned to each Class I ECP by the originator to indicate the urgency with which the ECP is to be reviewed, evaluated, ordered, and implemented. (The proposed priority as assigned and will stand unless the tasking activity has a valid reason for changing the priority.)
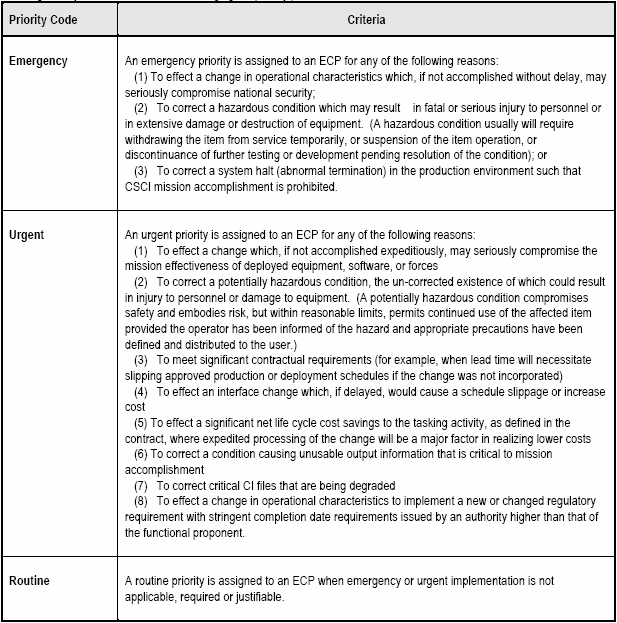
Table 6-6. Activity Guide: ECP Content
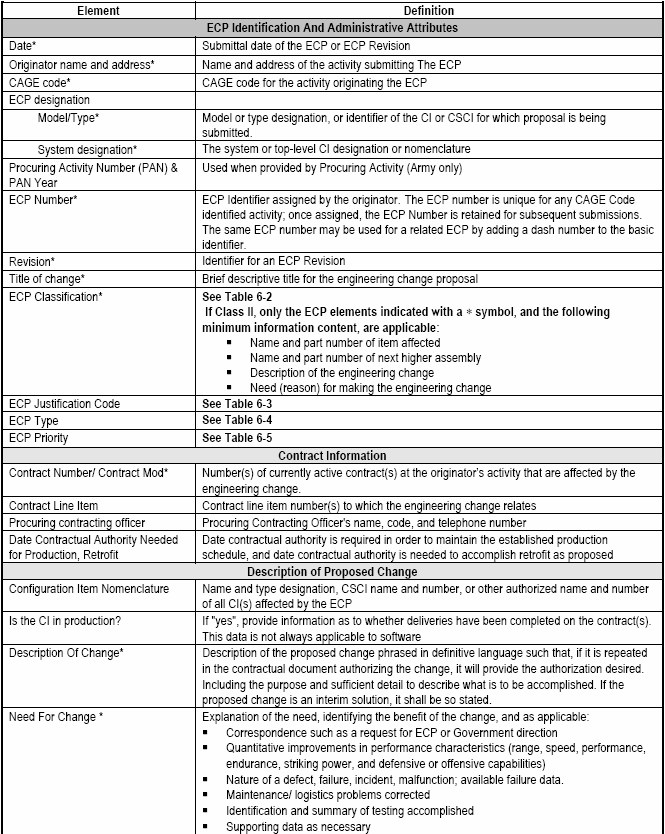
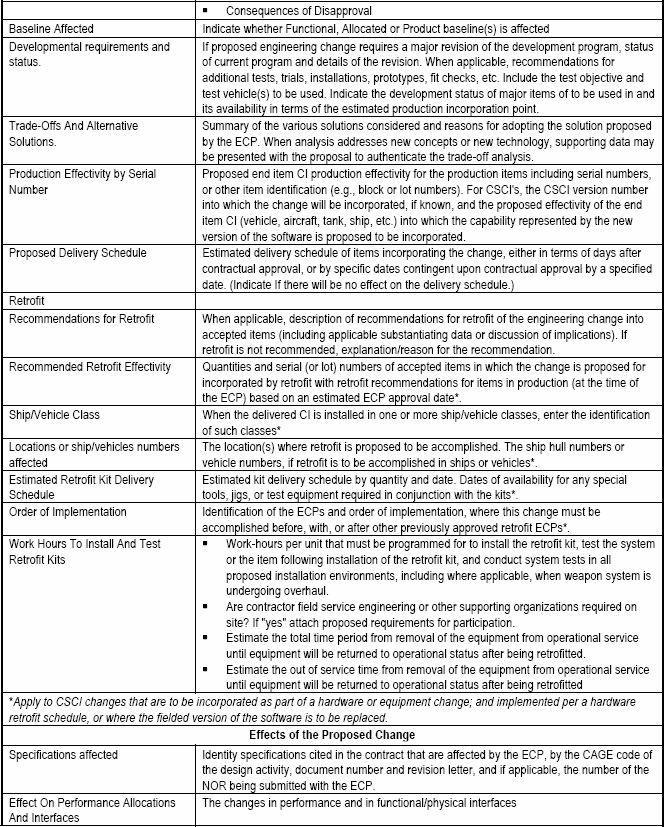

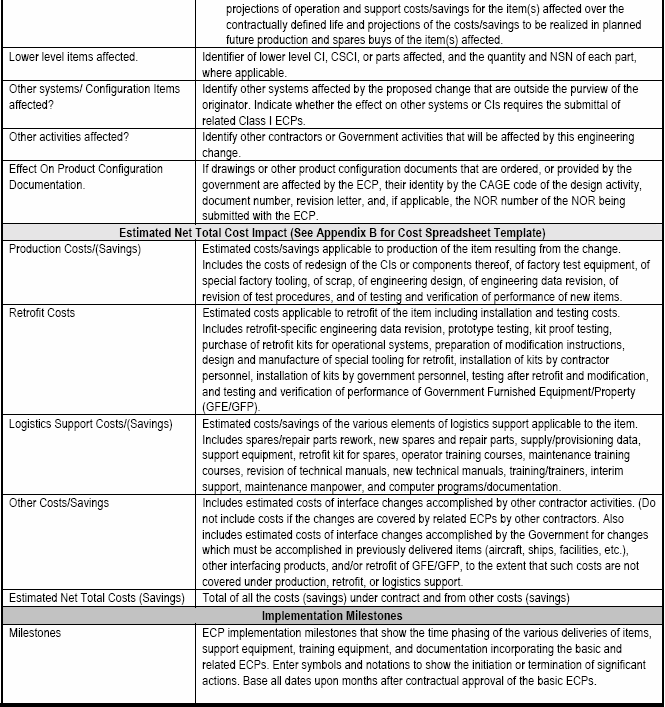
Table 6-7. Activity Guide: ECP Review and Disposition Actions
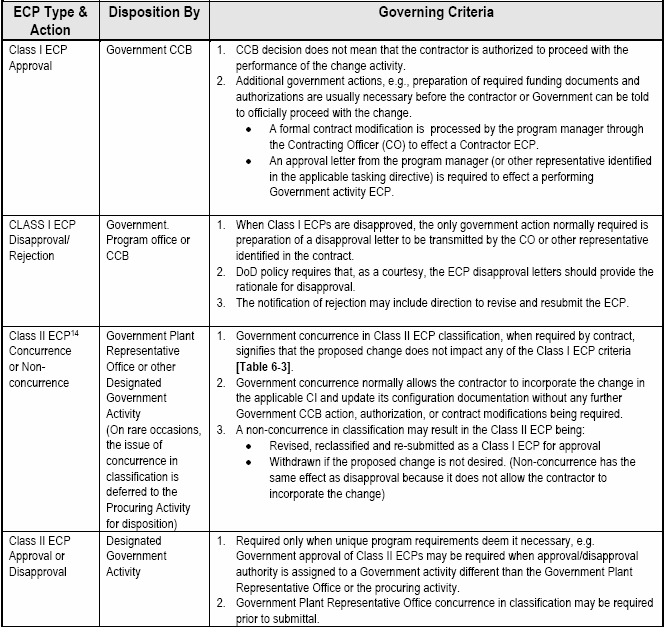
Table 6-8. Activity Guide: ECP Implementation Actions
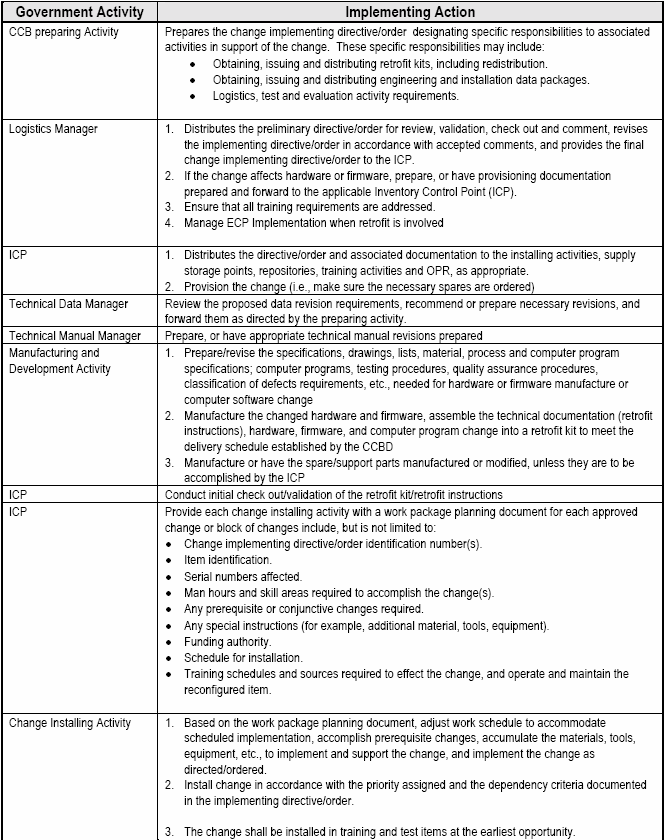
The OPR for MEARS is Commander, US Army MICOM, Attention: AMSMI-MMC-LS-SA (Mr. Mark Moe) Redstone Arsenal, Alabama, 35898-5238, DSN 746-9513
For correct application of this information, see NOTE on Contents page
 FAR
FAR
(a) Satisfy the customer in terms of cost, quality, and timeliness of the delivered product or service.
(1) The principal customers for the product or service provided by the System are the users and line managers, acting on behalf of the American taxpayer.
(2) The System must be responsive and adaptive to customer needs, concerns, and feedback. Implementation of acquisition policies and procedures, as well as consideration of timeliness, quality, and cost throughout the process, must take into account the perspective of the user of the product or service.
(3) When selecting contractors to provide products or perform services, the Government will use contractors who have a track record of successful past performance or who demonstrate a current superior ability to perform.
(4) The Government must not hesitate to communicate with the commercial sector as early as possible in the acquisition cycle to help the Government determine the capabilities available in the commercial marketplace. The Government will maximize its use of commercial products and services in meeting Government requirements.
(5) It is the policy of the System to promote competition in the acquisition process.
(6) The System must perform in a timely, high quality, and cost-effective manner.
(7) All members of the Team are required to employ planning as an integral part of the overall process of acquiring products or services. Although advance planning is required, each member of the Team must be flexible in order to accommodate changing or unforeseen mission needs. Planning is a tool for the accomplishment of tasks, and application of its discipline should be commensurate with the size and nature of a given task.
(b) Minimize administrative operating costs.
(1) In order to ensure that maximum efficiency is obtained, rules, regulations, and policies should be promulgated only when their benefits clearly exceed the costs of their development, implementation, administration, and enforcement. This applies to internal administrative processes, including reviews, and to rules and procedures applied to the contractor community.
(2) The System must provide uniformity where it contributes to efficiency or where fairness or predictability is essential. The System should also, however, encourage innovation, and local adaptation where uniformity is not essential.
(c) Conduct business with integrity, fairness, and openness.
(1) An essential consideration in every aspect of the System is maintaining the public’s trust. Not only must the System have integrity, but the actions of each member of the Team must reflect integrity, fairness, and openness. The foundation of integrity within the System is a competent, experienced, and well-trained, professional workforce. Accordingly, each member of the Team is responsible and accountable for the wise use of public resources as well as acting in a manner which maintains the public’s trust. Fairness and openness require open communication among team members, internal and external customers, and the public.
(2) To achieve efficient operations, the System must shift its focus from “risk avoidance” to one of “risk management.” The cost to the taxpayer of attempting to eliminate all risk is prohibitive. The Executive Branch will accept and manage the risk associated with empowering local procurement officials to take independent action based on their professional judgment.
(3) The Government shall exercise discretion, use sound business judgment, and comply with applicable laws and regulations in dealing with contractors and prospective contractors. All contractors and prospective contractors shall be treated fairly and impartially but need not be treated the same.
(d)Fulfill public policy objectives. The System must support the attainment of public policy goals adopted by the Congress and the President. In attaining these goals, and in its overall operations, the process shall ensure the efficient use of public resources.
What’s New in the 1 Form Proposal - Invoice 1.4 serial key or number?
Screen Shot
System Requirements for 1 Form Proposal - Invoice 1.4 serial key or number
- First, download the 1 Form Proposal - Invoice 1.4 serial key or number
-
You can download its setup from given links:

1 Form Proposal - Invoice 1.4 serial key or number & Key Download

1 Form Proposal - Invoice 1.4 serial key or number& Kali Software Crack
